Themenfocus
Prionen und ZNS-Erkrankungen |
||||||||||
| Dr. Karin Post und Prof. Dr. Detlev Riesner, Heinrich-Heine-Universität Düsseldorf Nicht zuletzt die Unklarheit über die Eigenschaften und die Übertragung des Erregers von Prion-Krankheiten macht die aktuelle Diskussion um BSE und der neuen Variante der Creutzfeld-Jakob-Krankheit brisant. In diesem Artikel fassen Dr. Post und Prof. Dr. Riesner den wissenschaftlichen Kenntnisstand bis Ende 1999 in verständlicher Weise zusammen. |
|
|||||||||
Bereits im
Jahre 1759 wurde von dem deutschen Veterinärmediziner Leopoldt die Traberkrankheit der
Schafe und Ziegen beschrieben. Im Angelsächsischen wird diese Krankheit nach dem
Kratzsymp-tom „Scrapie“ genannt. Sie kommt in vielen Ländern endemisch
vor. In den zwanziger Jahren dieses Jahrhunderts wurde von den deutschen rzten Creutzfeldt
und Jakob unabhängig voneinander eine menschliche Krankheit des zentralen Nervensystems,
die „Creutzfeldt-Jakob-Krankheit“ (CJD), beschrieben. Beide Krankheiten
beginnen nach einer langen Inkubation mit Bewegungsstörungen, führen zum totalen Verlust
der Koordinationsfähigkeit, zum Verfall der geistigen Kräfte und enden stets tödlich.
Die Symptome sind auf den Untergang von Neuronen und die Vermehrung von Gliazellen
(Astrozyten) zurückzuführen. Die Folge ist eine spongiforme, d.h. schwammartige
Veränderung des Gehirns, wie sie in Abb. 1 sichtbar ist.
Beide Krankheiten erwiesen sich als übertragbar. Man bezeichnet sie deshalb als transmissible
spongiforme Enzephalopathien (Tab. 1).
Abb. 1: Schnitt durch ein krankes
Gehirn (graue Gehirnsubstanz). |
Wirt |
Verbreitung und Entstehung |
|
| Scrapie | Schaf und Ziege | Infektion |
| Bovine spongiforme Enzephalopathie | Rind | Infektion |
| Creutzfeldt-Jakob-Erkrankung sporadisch | Mensch | Spontane PrPC zu PrPSc Umlagerung oder somatische Mutation im PrP-Gen |
| Creutzfeldt-Jakob-Erkrankung iatrogen | Mensch | Infektion |
| Creutzfeldt-Jakob-Erkrankung familiär | Mensch | Mutation im PrP-Gen |
| Creutzfeldt-Jakob-Erkrankung Variante | Mensch | Infektion durch BSE-Material |
| Gerstmann-Sträussler-Scheinker-Syndrom | Mensch | Mutation im PrP-Gen |
| Fatale Familiäre Insomnie | Mensch | Mutation im PrP-Gen Kuru Mensch Infektion |
Tabelle 1: Menschliche und tierische transmissible spongiforme Enzephalopathien
Der
Erreger
Die Frage nach der Natur des Erregers der Scrapie-Krankheit hat die Wissenschaft die letzten 30 - 40 Jahre beschäftigt und wird auch heute noch kontrovers diskutiert. Die vergleichsweise langen Inkubationszeiten bis zum Ausbruch der Krankheit führten zu dem Begriff „slow virus diseases“, der von Gajdusek geprägt wurde. Aber schon das Fehlen jeglicher Immunantwort sprach gegen einen Erreger vom Virustyp. Bereits 1966 endeckte die englische Strahlenbiologin Tikvah Alper die außerordentliche Resistenz des Scrapie-Erregers gegenüber Inaktivierung mit ionisierender und ultravioletter Strahlung und leitete daraus ab, dass der Erreger deutlich kleiner als ein Virus sein müsse und keine wesentlichen Nukleinsäure-Anteile enthalten könne. Diese Arbeiten wurden durch Stanley Prusiner systematisch fortgeführt und bestätigt. Er fand in langen Versuchsreihen, dass infektiöse Fraktionen durch Behandlung mit Nukleinsäure modifizierenden oder zerstörenden Agenzien nicht inaktiviert werden, wohingegen Proteinzerstörende oder verändernde Bedingungen einen Verlust der Infektiösität zur Folge hatten. Prusiner prägte daraufhin 1982 den Begriff des „Prion“ als proteinartiges infektiöses Partikel, welches ohne Nukleinsäure auszukommen scheint. Da Prionen an allen transmissiblen spongiformen Enzephalopathien beteiligt sind, werden diese Krankheiten mehr und mehr auch als Prion-Erkrankungen bezeichnet. Für das Prion-Modell, d.h. einen vollkommen neuartigen Erregertyp, erhielt Prusiner 1997 den Nobelpreis für Medizin und Physiologie. Die Annahme, dass ein Protein alleine als infektiöses, replizierendes Agens dienen soll, spricht gegen das zentrale Dogma der Molekularbiologie, wonach der Fluss der genetischen Information immer von der Nukleinsäure zum Protein verläuft. Bis heute konnten in infektiösen Präparationen keine Nukleinsäuren mit codierender Funktion identifiziert werden. Vielmehr konnte ausgeschlossen werden, dass Nukleinsäuren mit einer Größe über 50 Nukleotiden essentiell für die Infektiösität sind (Arbeitsgruppe Prof. Riesner).
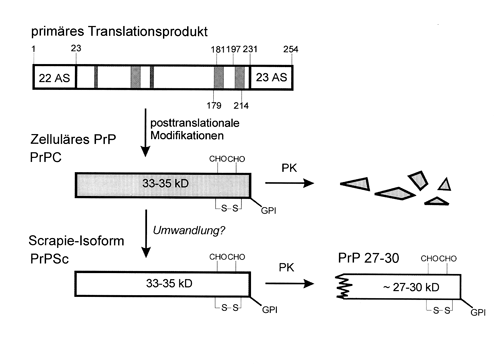
Als Hauptbestandteil infektiöser
Präparationen wurde ein Protein, das sogenannte Prion Protein (PrP), mit einem
Molekulargewicht von 33-35 kDa identifiziert. Durch aminoterminale
Aminosäuresequenzierung konnte die entsprechende cDNA identifiziert werden. Hierbei
stellte sich überraschenderweise heraus, dass es sich um ein wirtseigenes Gen und somit
nicht um ein exogenes Genprodukt handelte. Das bedeutet, dass auch der gesunde Organismus
das Prion-Protein produziert, allerdings als ungefährliches, sogenanntes zelluläres
Prion-Protein (PrPC). Die mit der Krankheit assoziierte Form hat ebenfalls eine Größe
von 33-35 kDa und wird als Scrapie-Form bzw. PrPSc bezeichnet. Während PrPC von Proteasen
leicht verdaut wird, wird von PrPSc nur ein N-terminales Peptid von 70 AS abgeschnitten,
und der Rest, das sogenannte PrP 27-30, ist gegen weitere Proteasen resistent (Abbildung 2). PrPC und PrPSc sind mit Zucker-Seitenketten und
Lipidanker hoch modifiziert. Bisher wurden weder chemische Unterschiede in der
Aminosäuresequenz noch in der Modifizierung gefunden. PrPC ist in Wasser -oder besser
milden Detergenzien - löslich; die pathogene Isoform PrPSc hingegen ist unlöslich.
Vergleichende spektroskopische Strukturanalysen zeigen jedoch einen erheblichen
Unterschied in der Sekundärstruktur. PrPC enthält einen relativ hohen a-helikalen Anteil und fast
keine b-Faltblatt-Elemente;
PrPSc hingegen weist mit einem erhöhten b-Faltblatt und geringeren a-Helix-Anteil eine andere
Sekundärstrukturverteilung auf. Demnach handelt es sich bei PrPC und PrPSc um
Strukturisomere. Dabei scheint PrPSc posttranslational aus PrPC gebildet zu werden.
PrPSc akkumuliert während des Krankheitsverlaufes und bildet Plaques und Ablagerungen im
Hirn betroffener Individuen. Die dreidimensionale Struktur des PrPC konnte inzwischen mit
der Methode der NMR aufgeklärt werden.
| Abb. 2: Biosynthese von PrPC und PrPSc. Die reifung des PrP?Vorläuferproteins beinhaltet die Abspaltung der N?terminalen Signalsequenz (siehe Artikel auf S. 6f), das Anhängen des Glykolipidankers (GPI) an Aminosäure (AS) 231 nach Entfernung der C?terminalen Signalsequenz, der Glykosylierung an den Positionen 181 und 197 sowie der Ausbildung einer Disulfidbrücke zwischen AS 179 und 214. Das "gereifte" PrPC ist sensitiv gegenüber Verdauung mit Proteinase K (PK). (Zeichnung verändert nach Weismann, 1994) |
Die zelluläre Funktion hingegen ist noch weitgehend unverstanden. PrPC scheint eine Rolle bei der synaptischen Őbertragung und der Regulation von zirkadianer Aktivität (Chronobiologie; siehe Biologen heute 4//99) zu spielen. Außerdem wurden kürzlich kupferbindende Eigenschaften des Proteins beschrieben, deren Bedeutung für die krankheitsassoziierten Symptome aber noch unklar ist. Bei einigen anderen ZNS-Krankheiten sind aber ebenfalls Cu bindende Proteine beteiligt (z.B. Alzheimer, Parkinson).
Ein Modell zur Vermehrung von Prionen muss das Auftreten von spontan auftretenden (sporadischen), übertragbaren (infektiösen) und familiären (genetisch bedingten) Formen von Prionerkrankungen erklären können. Außerdem muss die Existenz verschiedener Scrapie-Stämme erklärbar sein. Letztere werden oft als Argument für eine Scrapie-spezifische Nukleinsäure und damit für die Virushypothese angeführt; so induzieren unterschiedliche Scrapie-Isolate in Schafen mit identischem genetischen Hintergrund verschieden lange Inkubationszeiten und unterschiedliche neuropathologische Läsionsmuster.
Charles Weismann und Stanley Prusiner publizierten 1992
transgene Mäuse, denen das Gen für das Prion-Protein gentechnisch entfernt wurde,
sogenannte Knock-out-Mäuse. Nach Inokulation mit infektiösem Material wurden
diese weder krank noch konnten sie die Infektion weiterreichen. Folglich ist das
körpereigene Protein absolut notwendig für den Ausbruch der Krankheit; ob es auch
hinreichend ist bleibt noch zu klären. Die Theorie der „Protein-only“-Hypothese,
wie sie von Prusiner formuliert wurde, beinhaltet, dass die Konformationsänderung
von der zellulären, gesunden in die pathogene Struktur der relevante Schritt für die
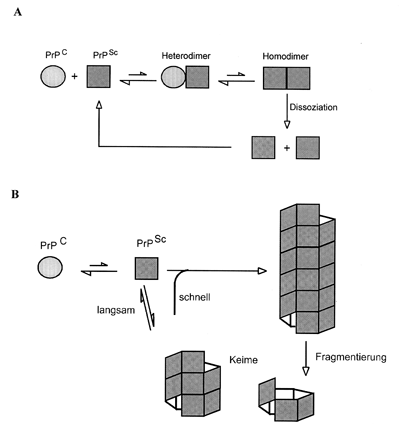
Prion-Vermehrung ist. Das sogenannte Heterodimer-Modell (Abbildung
3) besagt, dass ein PrPSc-Molekül durch direkten Kontakt die Umlagerung eines
PrPC-Moleküls induzieren kann. Dabei wird zunächst ein Heterodimer ausgebildet, das dann
in ein Homodimer umgelagert wird. Dieses Homodimer dissoziiert und zwei PrPSc-Moleküle
stehen für den nächsten Zyklus zur Verfügung. Sporadische Fälle wären somit auf eine
spontane Umlagerung von PrPC zur PrPSc-Form zurückzuführen oder auf eine somatische
Mutation im PrP-Gen. Durch eine Veränderung in der PrP-Sequenz, wie es bei familiären
Creutzfeldt-Jakob-Erkrankungen der Fall ist, würde die Wahrscheinlichkeit für eine
spontane Umlagerung aufgrund der geringeren thermodynamischen Stabilität des mutanten
PrPC-Moleküls steigen. Verschiedene Stämme wären viele verschiedene pathogene
Strukturisomere von PrPSc. Eine Alternative zum Heterodimer-Modell ist das von Lansbury
vorgeschlagene keimabhängige Polymerisationsmodell (Abbildung 3).
Hierbei ist die Umlagerung von PrPCzu PrPSc mit einer Anlagerung an PrPSc-Aggregate
verbunden. Das entstehende PrPSc-Molekül ist alleine nicht stabil, sondern wird durch die
Anlagerung an bereits bestehende Aggregate stabilisiert. Diese Polymere können wieder
zerfallen und somit die Anzahl aktiver Keime erhöhen.
| Abb. 3: A) Heterodimer-Modell. Ein monomeres PrPSc katalysiert die Konformationsumwandlung von PrPC in PrPSc über einen heterodimeren Zwischenzustand. B) Keim?abhängiges Polymerisationsmodell. Monomeres PrPC und PrPSc stehen in einem schnellen Gleichgewicht, wobei die PrPSc-Isoform durch die Anlagerung an bereits bestehende PK-resistente Aggregate stabilisiert und dem Gleichgewicht entzogen wird. Durch Fragmentierung bereits bestehender größerer PrPSc-Aggregate wird die Anzahl der Keime erhöht. |
Die In vitro-Konversion, dass heißt die Umwandlung von PrPC zu PrPSc im Reagenzglas, wäre der letztendliche Beweis der „Protein-only“-Hypothese. Trotz intensiver Forschung vieler Gruppen ist es bis jetzt nicht gelungen Infektiösität de novo zu synthetisieren. Daher kann ein Einfluss weiterer Faktoren wie Proteine, Lipide oder Kohlenhydrate auf die Infektiösität oder Pathogenität von Prionen nicht letztendlich ausgeschlossen werden. Für zusätzliche Faktoren spricht auch, dass Mäuse nicht oder nur mit langen Inkubationszeiten mit menschlichen Prionen infiziert werden können. Mäuse, die transgen für das menschliche PrP sind, sind ebenfalls nicht oder kaum mit menschlichen Prionen infizierbar. Hingegen sind transgene Mäuse, bei denen nur ein Teil des Maus-PrP durch menschliche PrP-Sequenzen ersetzt wurde (sogenantes chimäres Protein), mit menschlichen Prionen infizierbar. Dies lässt vermuten, dass noch nicht weiter identifizierte und daher mit Protein X bezeichnete Faktoren an der Vermehrung des Prions beteiligt sind.
Die Creutzfeldt-Jakob-Erkrankung ist mit einer Häufigkeit von einem Fall unter 1 Mio. Einwohnern pro Jahr selten, aber weltweit vertreten. Die Krankheit kann sowohl spontan als auch infektiös oder genetisch bedingt auftreten, was vorher bei keiner anderen Krankheit bekannt war. Etwa 85% der CJD-Fälle sind sporadischer Natur, der Rest ist familären Ursprungs. Letztere sind ebenso wie die Fatale Familiäre Insomnie (FFI) und das Gerstmann-Sträussler-Scheinker- Syn-drom (GSS) auf Mutationen im PrP-Gen zurückzuführen. Die Ursachen für die seltenen infektiösen Őbertragungen der Krankheit lagen in unzureichend sterilisierten Instrumenten bei neurochirurgischen Eingriffen oder bei Dura-mater- und Korneatransplanta
tionen von unerkannten Krankheitsträgern (sog. iatrogene Őbertragungen). Außerdem sind weltweit über 40 Patienten an CJD erkrankt, die mit kontaminierten Wachstumshormonen aus menschlichen Hypophysen behandelt wurden. Ebenfalls als infektiöse Prionerkrankung gilt Kuru; eine humane Erkrankung, die vor allem Kinder und Frauen des Fore-Stammes in Papua-Neuguinea betrafen. Die Őbertragung erfolgte hierbei durch einen Ritus, bei dem vorwiegend die Frauen die Gehirne verstorbener Angehöriger präpariert haben und sich dabei ansteckten. Seit dem Verzicht auf diese rituellen Handlungen sind keine weiteren Neuinfektionen mehr aufgetreten.
Sowohl epidemiologisch als auch
ökonomisch von besonderer Bedeutung ist die seit 1985 in Großbritannien auftretende
bovine spongiforme Enzephalopathie (BSE), die im Volksmund als „Rinderwahnsinn“
bekannt ist. Als Ursache gilt die Verfütterung von Tierkörpermehl an Rinder, welches
Kadaver von Scrapie-infizierten Schafen enthielt. Die Herstellungsverfahren für
Tierkörpermehl wurden Ende der 70er Jahre auf niedrigere Temperaturen umgestellt und es
wurde auf zusätzliche Extraktionsverfahren verzichtet. Dadurch wurde der Scrapie-Erreger
nicht mehr hinreichend desaktiviert. Seit 1988 darf daher kein Tiermehl mehr an
Wiederkäuer verfüttert werden und seit 1989 dürfen keine bovinen Innereien mehr in die
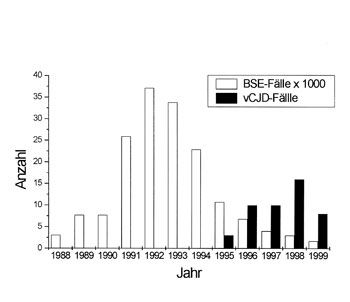
menschliche Nahrungskette gelangen. Im Jahre 1992 erreichte die Epidemie in
Großbritannien mit knapp 37.000 Fällen/Jahr ihren Höhepunkt (Abbildung
4).
|
Abb. 4: Anzahl der BSE-Fälle (x
1000!) |
Bis Juli 1999 waren insgesamt 177.000 Rinder betroffen und derzeit erkranken etwa noch 100 - 200 Tiere monatlich. Die Inkubationszeit für BSE beträgt im Schnitt 3 -5 Jahre. Neben Großbritannien sind vor allem die Schweiz und Irland betroffen. In Zusammenhang mit dem Auftreten von BSE wurden auch bei verschiedenen Zootieren Prionerkrankungen beobachtet, die wahrscheinlich ebenfalls auf die Verfütterung von kontaminiertem Tierkörpermehl zurückzuführen sind.
Nachdem wahrscheinlich mehr als 1 Million BSE-infizierter Rinder in die menschliche Nahrungskette gelangt sind, wird nun eine weitere menschliche Krankheit ursächlich mit dem Auftreten von BSE in Zusammenhang gebracht. 1996 wurde eine neue Variante der menschlichen CJD (vCJD) beschrieben, an der bisher insgesamt 48 Patienten (Stand Sep. 1999), 46 davon in Großbritannien, erkrankten (Abbildung 4). Im Vergleich zur herkömmlichen CJD, mit einem durchschnittlichen Erkrankungsalter von 65 Jahren, liegt das Erkrankungsalter bei der neuen Variante im Schnitt bei 27 (16-48) Jahren. Die jungen Patienten fallen zunächst durch Verhaltensveränderungen, wie gesteigerte Aggressivität, auf. Der klinische Krankheitsverlauf dauert mit durchschnittlich 16 (9-38) Monaten etwa doppelt so lang, wie bei sporadischer CJD. Tabelle 1 zeigt eine Zusammenstellung der transmissiblen spongiformen Enzephalopathien (TSE).
Eine Reihe von histopathologischen und biochemischen Befunden unterstreichen, dass die vCJD eher Eigenschaften des BSE-Erreger, als von sporadischer CJD zeigt. Makaken, die mit BSE infiziert wurden, zeigten ähnliche Läsionsmuster im Gehirn wie Patienten, die an vCJD erkrankten. Darüber hinaus waren die Glykosylierungsmuster der Erreger von BSE und vCJD identisch. Daher ist der Verzehr von mit BSE kontaminierten Nahrungsmitteln als Ursache für die vCJD nicht mehr auszuschließen. Infektionstests bei Mäusen zeigten keine nachweisbare Infektiösität in Muskelfleisch oder Milch der Rinder. Infektiösität wurde in verschiedenen Geweben des Nervensystems, wie in Ganglien, der Retina und im Knochenmark, gefunden. Daraufhin wurde 1998 in Großbritannien der Verkauf von „beef on the bone“ verboten. Das lymphatische System spielt, neben der direkten Invasion des Erregers über das Nervensystem, eine zentrale Rolle bei der Verbreitung des Erregers im Körper. B-Zellen transportieren die Infektiösität, die zunächst in der Milz akkumuliert und später auch in den Mandeln zu finden ist. Daher werden in Großbritannien seit 1998 Blutkonserven depletiert, d.h. die Leukozyten als mögliche Träger des infektiösen Agens werden entfernt. Inzwischen konnte der Erreger mit molekularbiologischen Methoden auch in den Mandeln und im Wurmfortsatz des Blinddarms von vCJD-Patienten nachgewiesen werden. Diese Gewebe sind derzeit Gegenstand anonymer Studien, um den Durchseuchungsgrad für vCJD in der britischen Bevölkerung festzustellen. Alle derzeit in Anwendung befindlichen BSE-Tests beruhen auf dem Post-mortem-Nachweis des proteaseressistenten Prion-Proteins in Hirngewebe von Rindern. Testverfahren am lebenden Tier befinden sich noch in der Entwicklungsphase. Hoffnung gibt hier jedoch ein Diagnostiksystem, welches inzwischen erfolgreich bei der Alzheimerschen Krankheit angewendet wird. Bei dieser Krankheit kommt es zur Ablagerung von Peptidaggregaten im Hirn (s. unten). In unserem Institut ist es gelungen, diese Aggregate mit Hilfe der Fluoreszenz-Korrelations-Spektroskopie in der Rückenmarksflüssigkeit von Alzheimer-Patienten spezifisch nachzuweisen.
Fehlerhafte Proteinstruktur löst verschiedene Krankheiten aus
Im Fall der Prionkrankheiten liegt der pathologische Effekt nicht darin, dass die Funktion eines lebenswichtigen Proteins ge- oder zerstört ist (das ist zumindest die Vermutung nach den Ergebnissen des „Knock-out-Experimentes“), sondern das falsch gefaltete Protein ist so stabil, dass es Aggregatsablagerungen ausbildet und so seine schädliche Wirkung ausübt.
Eine ganze Reihe von Erkrankungen gehen mit einer veränderten und damit pathogenen Form von körpereigenen Proteinen einher. In allen Fällen ist eine Abnahme von a-helikalen Strukturbereichen zugunsten einer Zunahme an b-Faltblatt-Strukturelementen zu beobachten. Dies führt zur Ausbildung sogenannter amyloider Aggregate, die im Gewebe abgelagert werden. Allgemein werden diese Krankheiten als Amyloidosen zusammengefasst. Die amyloiden Fibrillen können aus den amyloiden Proteinen selbst (z.B. Lysozym) oder aus durch Proteasen verkürzten Untereinheiten bestehen (z.B. Amyloid-b-Peptid). Zu den Amyloidosen gehören u.a. die familären amyloidischen Polyneuropathie und die Alzheimersche Krankheit. Im Gegensatz zu den Prionerkrankungen treten diese jedoch nicht als infektiöse Formen auf.
Informationen zu Prionen/BSE/CJD im Internet:
Eine kleine Auswahl, dort weitere "Links":
| Dieser Artikel wurde der Zeitschrift
"Biologen heute", Ausgabe 6/1999 entnommen. |
|
[Eintragen in Mailingliste] [Bookmark: Bitte hier rechts klicken und "hinzufügen" wählen] |
||
| www.BioloGE.de
- Letzte Änderung: 29-Nov-2000 Diese Sektion wird betreut von wiegand@biologe.de ©BioloGE.de - |
||